FDA courses in Bristol
We couldn't find any listings for your search. Explore our online options below.
Know someone teaching this? Help them become an Educator on Cademy.
Get a 10% discount on your first order when you use this promo code at checkout: MAY24BAN3X
Online Options
Show all 17CT10: The Investigational New Drug Application (IND) to Conduct FDA-regulated Clinical Trials
By Zenosis
An Investigational New Drug Application (IND) is a submission to the US Food and Drug Administration (FDA) for permission to conduct a clinical trial of a medicinal product. This module describes regulatory requirements that sponsors or sponsor-investigators must meet for successful compilation, filing and maintenance of INDs. The IND and its role are defined, and the contexts in which it is required are specified.
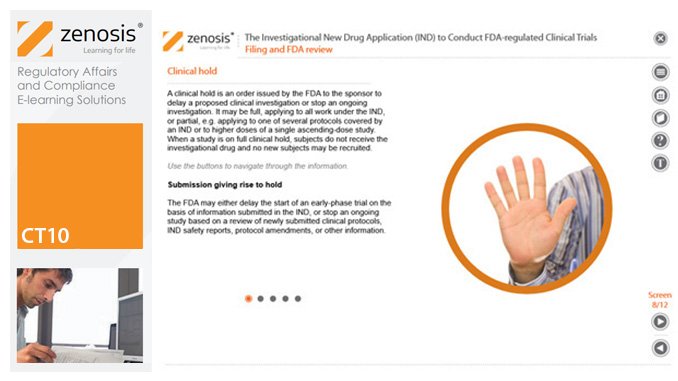
ICT01: Compliance with Regulation 21 CFR Part 11 on Electronic Records and Electronic Signatures
By Zenosis
21CFR11 applies to records that are required to be submitted to the FDA, or that are subject to FDA inspection, and that are in electronic form – that is, as computer files. It applies to all computer systems used to create, modify, maintain, archive, retrieve, or transmit such records – from a humble spreadsheet program to a complex information management system.
SUB09: The New Drug Application (NDA) for Marketing Approval in the USA
By Zenosis
The New Drug Application (NDA) is the regulatory vehicle through which sponsors formally propose that the Food and Drug Administration (FDA) approve a new pharmaceutical for marketing and sale in the USA.
SAM02: Regulatory Requirements and Guidance on Advertising and Promotion of Prescription Drugs in the USA
By Zenosis
In this course we explain how to advertise and promote prescription drugs in various media, whether to healthcare professionals or consumers, in compliance with legal requirements and guidance from the FDA.

SUB14: The Regulatory Pathway to Licensure of Follow-on Biologics (Biosimilars) in the USA
By Zenosis
The regulation of biological medicinal products is governed by different laws from those that apply to small-molecule synthetic drugs. Producing faithful copies of therapeutic proteins is more challenging than producing generic drugs. The US legal framework for the licensure of follow-on biologics, and accompanying regulatory guidance from the Food and Drug Administration (FDA), have been established only in recent years.

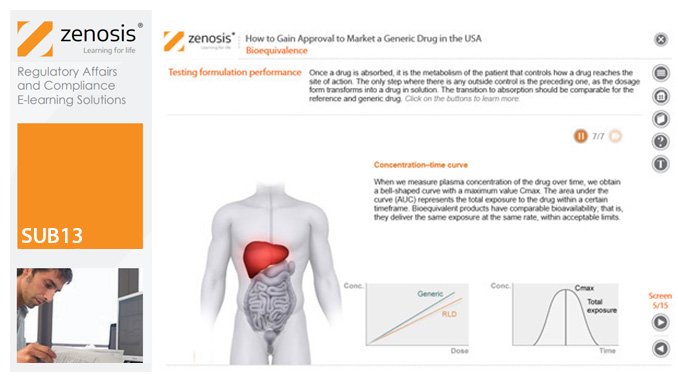
SUB13: How to Gain Approval to Market a Generic Drug in the USA
By Zenosis
This module outlines the legislative and regulatory context for the development of generic drugs and describes the essential role of the Abbreviated New Drug Application (ANDA) in gaining marketing approval. The use of information in the ‘Orange Book’ is explained, as is the role of patent certification in the application. The importance of establishing bioequivalence between a generic and its reference product is emphasised. The module specifies the content and format requirements for an ANDA submission and describes the FDA’s review and approval process. An outline is given of the Generic Drug User Fee Amendments (GDUFA) and the law’s effects on industry players.
CT09: Good Clinical Practice Inspections and Audits
By Zenosis
The module describes general principles of GCP inspection and audit, discusses preparation for an inspection, and sets out in detail what European and US FDA inspectors will examine. Finally it describes post-inspection actions by the regulator and the inspected party.

SUB15: The Biologics License Application (BLA) for Marketing Approval in the USA
By Zenosis
This module describes the requirements that must be met to obtain licensure of a biological product. Subjects covered include the regulatory context, the content and format of the BLA submission, the review process, and provisions for expedited development and review.


