- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
10908 Professional Development courses delivered On Demand
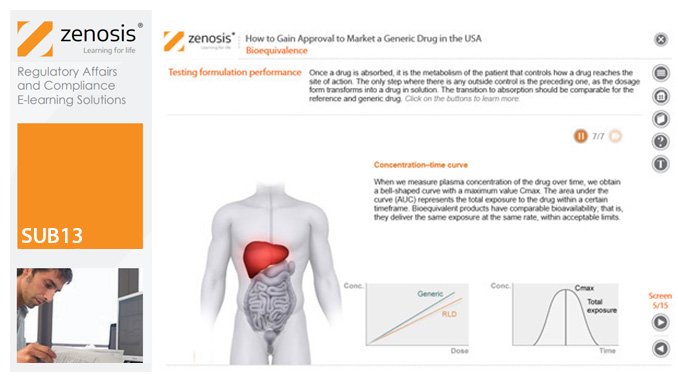
SUB13: How to Gain Approval to Market a Generic Drug in the USA
By Zenosis
This module outlines the legislative and regulatory context for the development of generic drugs and describes the essential role of the Abbreviated New Drug Application (ANDA) in gaining marketing approval. The use of information in the ‘Orange Book’ is explained, as is the role of patent certification in the application. The importance of establishing bioequivalence between a generic and its reference product is emphasised. The module specifies the content and format requirements for an ANDA submission and describes the FDA’s review and approval process. An outline is given of the Generic Drug User Fee Amendments (GDUFA) and the law’s effects on industry players.
VAL07: Computer Systems Validation, Part 2: Implementation
By Zenosis
This module describes the design, development and installation phase, the validation phase, and the operation and maintenance phase of the validation of computerised systems in medicines and healthcare products manufacturing environments. It continues to follow the progress of a pharmaceutical company's project to validate a new dispensary control system.
VAL03: Commissioning and Installation Qualification
By Zenosis
Before equipment can be used routinely in production, it must first be commissioned and, if necessary, undergo Installation Qualification (IQ). This module describes commissioning and IQ requirements and procedures in the medicines and healthcare products industries. It follows the activities of a typical validation team as they carry out a project for a pharmaceutical company.
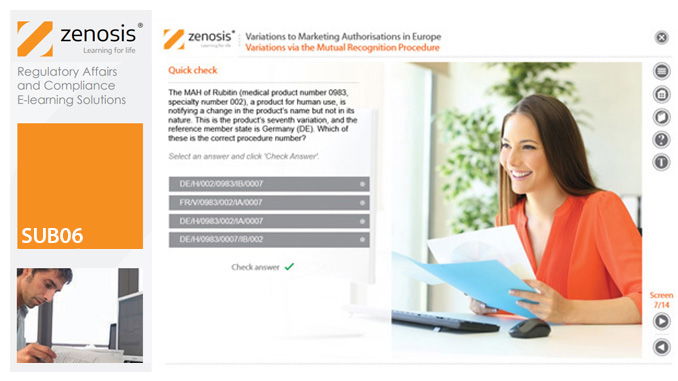
SUB06: Variations to Marketing Authorisations in Europe
By Zenosis
Changes to the terms of marketing authorisations for medicinal products, called variations in Europe, must be notified to or approved by the relevant regulatory authorities. Variations include changes to the composition of products, their manufacturing processes, the way they are used, or the indications for which they are authorised. Common approaches are adopted within the European Economic Area to variations to marketing authorisations approved through the Centralised, Decentralised or Mutual Recognition Procedures. Recent legislation has substantially modified the regulatory requirements and extended them to purely national authorisations by member states. This module, which is fully up to date with the new legislation, covers the classification of variations into their several types and the regulatory requirements, guidance and procedures to be followed for each type.
Sales, Marketing, Negotiation, Sales Funnels, Sales Analysis & Social Media - 20 Courses Bundle
By NextGen Learning

ANALYST ACADEMY PRO
By Behind The Balance Sheet
Join the Analyst Academy Pro and get a job as a job as a research analyst or simply fast-track your career in asset management.

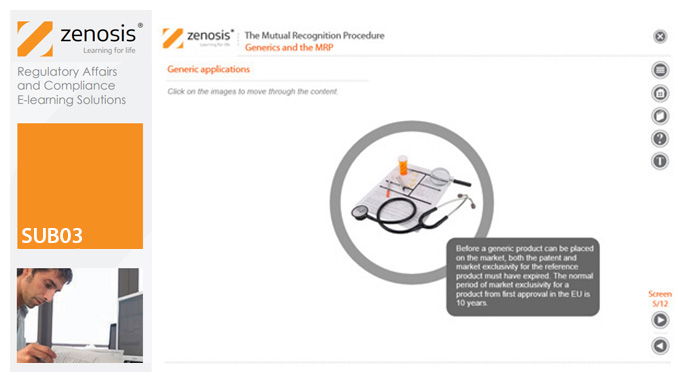
SUB03: The Mutual Recognition Procedure (MRP)
By Zenosis
his module describes the roles of the various players in the procedure, the sequence and duration of the stages involved, and the requirements on content, format and timing of submissions. It discusses the special issues that apply to generic products in the MRP.
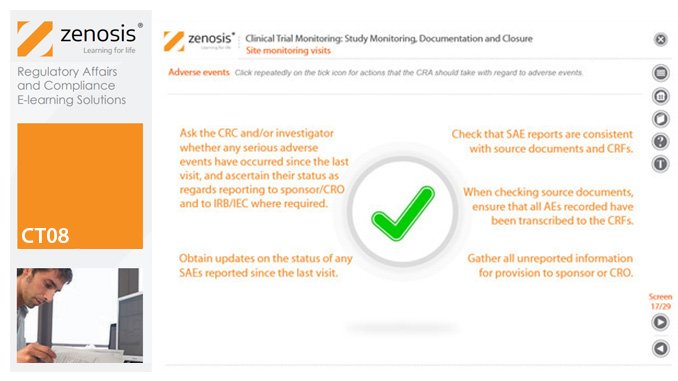
CT08: Clinical Trial Monitoring: Study Monitoring, Documentation and Closure
By Zenosis
The sponsor of a clinical trial must arrange for it to be monitored throughout its duration to ensure that the rights and wellbeing of subjects are protected, the trial data are accurate, complete and verified from source documents, and the conduct of the trial complies with the study protocol, Good Clinical Practice and regulatory requirements. In this module we describe how a Clinical Research Associate (CRA) monitors an ongoing trial to its conclusion.
SUB11: The Decentralised Procedure (DCP)
By Zenosis
This module describes the roles of the various players in the procedure, the sequence and duration of the stages involved, and the requirements on content, format and timing of submissions. It discusses the special issues that apply to generic products in the DCP.