- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
143 Courses
SUB04: Preparing Submissions in the Common Technical Document (CTD) Format
By Zenosis
The CTD is the internationally recognised standard format for submissions to medicines regulatory authorities. In the European Economic Area, the USA and Canada, the CTD, in its electronic format (eCTD), is mandatory for all applications for marketing approval and all subsequent related submissions. The CTD is accepted in many other countries, being mandatory for new prescription medicines in some. This module explains the rationale for the CTD and provides guidance on its structure and format and the ways in which it is used.

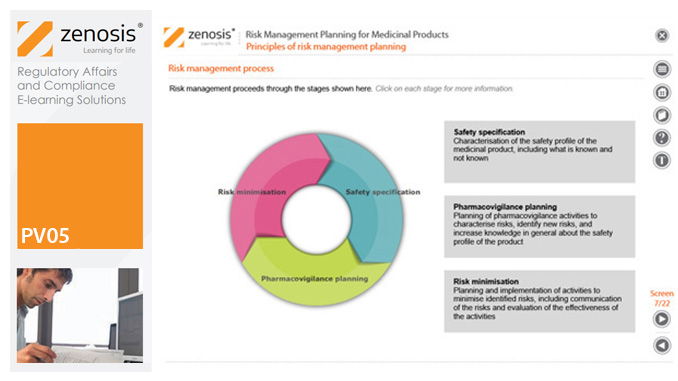
PV05: Risk Management Planning for Medicinal Products
By Zenosis
Proactive risk management is a major component of good pharmacovigilance practice. This module sets out the principles of risk management planning and outlines regulatory requirements for risk management plans in regions that are major markets for medicinal products.
SUB16: The 505(b)(2) Application for Marketing Approval in the USA
By Zenosis
A 505(b)(2) New Drug Application (NDA) is a submission to the Food and Drug Administration (FDA) for approval to market a drug in the USA. It differs from a ‘stand-alone’ NDA in that some of the data on which the applicant relies to demonstrate safety and efficacy have been obtained from publicly available sources rather than from the applicant’s own studies. The applicant typically proposes to market a drug that is based on an approved reference product but modified in its formulation or uses. A 505(b)(2) NDA also differs from an Abbreviated New Drug Application (ANDA) for approval of a generic drug in that the applicant’s product need not be a duplicate of the reference listed drug. The 505(b)(2) pathway may be said to lie part-way between the ‘stand-alone’ NDA and generics pathways, offering a unique combination of advantages to developers. It facilitates the modification of drugs to address unmet medical needs. The 505(b)(2) application pathway accounts for about half of all new drug approvals in the USA.

CT12: How to Conduct Clinical Research Under the EU Clinical Trials Regulation
By Zenosis
This course describes the requirements that must be met by, and options available to, the sponsor during the conduct of an authorised clinical trial. It identifies the various interactions with MSCs that occur via the Clinical Trials Information System (CTIS), and it summarises and links to the extensive guidance available from the European Commission and the European Medicines Agency. Its companion course CT11 sets out the European legal and regulatory context for clinical trials and describes how to apply via the CTIS for authorisation to conduct trials. The two courses therefore provide an ideal foundation for understanding and complying with the new law.

MD01: An Introduction to the Regulation of Medical Devices
By Zenosis
This module provides an introduction to the basics of medical device regulation, especially the requirements that manufacturers must meet in order to market devices in Europe and the USA.

ESS02: Essentials of Monoclonal Antibodies
By Zenosis
This module will introduce you to monoclonal antibodies, explaining how they work, how they are made, and the many uses to which they are put.

PV07: Good Pharmacoepidemiology Practice
By Zenosis
Pharmacoepidemiology is the study of the use and effects of drugs in large numbers of people. It provides a bridge between clinical pharmacology and epidemiology. The increasing demand for real-world evidence of the safety, efficacy and utility of medicinal products has focused greater attention on pharmacoepidemiological research. This module will help those who plan and conduct such research, and analyse and report the findings, to follow good practice.

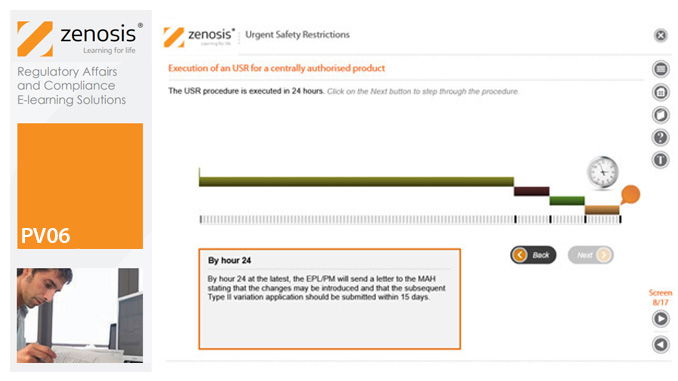
PV06: Urgent Safety Restrictions
By Zenosis
An Urgent Safety Restriction (USR) is a regulatory action taken, in response to a safety signal, to make an interim change to the terms of the marketing authorisation for a medicinal product in Europe. This module describes the principles and procedures for USRs.
CT04b - Clinical protocol design
By Zenosis
Clinical trial protocols are an essential part of clinical trial design. Protocol documents are critical to conducting safe and cost-effective investigations. Protocol documents are large and complex, containing comprehensive information relating to purpose, design and conduct of a clinical trial. Aspects of a protocol include patient eligibility criteria, and treatment specifications. This short course provides an overview of clinical trial protocols. Opportunities to improve a clinical trial protocol for regulatory approval are also discussed.
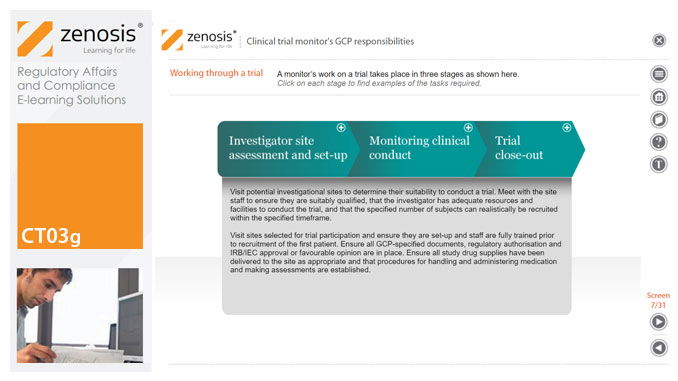
CT03g - Clinical trial monitor’s GCP responsibilities
By Zenosis
A clinical trial monitor acts on behalf of the sponsor to support investigational site personnel, verify the accuracy of data recorded, and ensure that the trial is conducted in compliance with the protocol, GCP and other study specific requirements. He or she acts as the ‘eyes and ears’ of the sponsor at the investigational site and provides the main channel of communication between sponsor and investigator. This short course explores the responsibilities of the monitor and provides insight into key challenges. We discuss assessment of investigators and investigational sites, education and trial initiation, monitoring of clinical conduct, including CRF review and source document verification, and trial close-out. We discuss noncompliance and how to deal with it.