- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
45 Courses delivered Online
CT14: Clinical Trial Safety Reporting Requirements in the EU and USA
By Zenosis
This course sets out the legal and regulatory requirements for safety reporting in clinical trials of medicinal products under the jurisdictions of the European Union and the USA. It builds on the foundation laid by our companion course CT13, Safety Reporting in Clinical Trials, and provides greater detail of specific requirements in those jurisdictions.

CT03: ICH Good Clinical Practice
By Zenosis
Good Clinical Practice (GCP) is a set of internationally recognised ethical and scientific quality requirements for designing, conducting, recording and reporting clinical trials. Compliance with GCP principles is required by regulatory authorities in many countries for the authorisation of clinical trials and the acceptance of their data. The International Council for Harmonisation’s guideline E6, often referred to as ICH GCP, is the international standard specification for Good Clinical Practice.

CT04c - Clinical trial preparation
By Zenosis
The demands on quality from clinical trials are increasing. Quantitative aspects of clinical trials, such as the mass of study data to be collected, the multiple investigational sites, and the need to meet predetermined timelines, often supersede qualitative features. Therefore, addressing basic requirements for quality management is essential when preparing a clinical trial. This short course describes the core elements required for the establishment of a clinical trial and provides an overview of the role of the sponsor in supporting and improving trial quality.
CT03a - ICH, harmonisation, and principles of Good Clinical Practice
By Zenosis
Good Clinical Practice (GCP) is a set of internationally recognised ethical and scientific quality requirements for designing, conducting, recording and reporting clinical trials. Compliance with GCP principles is required by regulatory authorities in many countries for the authorisation of clinical trials and the acceptance of their data in applications for marketing approval. The International Council for Harmonisation's guideline E6, often referred to as ICH GCP, is the international standard specification for Good Clinical Practice. In this short course we describe the ICH’s role in the harmonisation of regulations, introduce its guideline E6, and set out the principles of GCP.
CT04: An Introduction to Clinical Trial Preparation and Design
By Zenosis
This module aims to provide you with effective strategies for the preparation and conduct of a clinical trial, while adhering to regulatory safety standards. Management of data for submission is also covered.

CT04d - Clinical trial endpoints
By Zenosis
In clinical trials, endpoints are measurements to evaluate the results of a new treatment, at an individual patient level. The study data can be extrapolated to patient populations on the basis of clinical similarities to patients participating in the trial. When clinical trial data have been obtained, focus is on the trial endpoints; more specifically, the focus is on whether the trial met or failed the primary endpoint specified before the trial started. The purpose and various types of endpoints are discussed in this short course.
The Simplest Guide™ to Clinical Data Analysis with SAS
By Packt
Data science is quickly taking over all aspects of life, and a huge impact of this can be seen in the healthcare department. From medical imaging to genomics, we now leverage data to make better medical decisions. In this course, we will see how clinical trial data can be effectively managed using SAS.

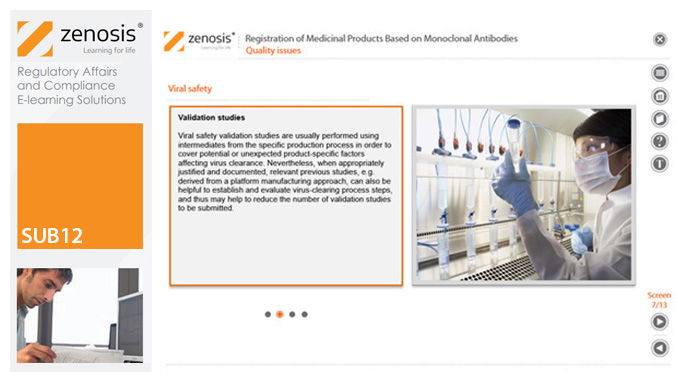
SUB12: Registration of Medicinal Products Based on Monoclonal Antibodies
By Zenosis
This module addresses characteristic issues influencing the registration of medicinal products based on monoclonal antibodies (mAbs), for use in humans. Regulatory requirements for the registration of biological medicinal products such as those based on mAbs differ in certain respects from those for small-molecule products. This is because of the distinct characteristics of biologics, such as complex structure and susceptibility to variation during manufacture.